
Trends In FDA FY 2024 Inspection-Based Warning Letters
By Liz Oestreich, Kalah Auchincloss, and Erin Hartmann, Eliquent Life Sciences

The U.S. FDA issued a total of 190 warning letters to drug and biologics manufacturers in Fiscal Year 2024 (FY24). Of those, 113 were based on an FDA inspection, including 12 letters issued to clinical investigators or sponsors following inspections conducted as part of the FDA’s Bioresearch Monitoring (BIMO) Program. For the purposes of this article, we have chosen to analyze 111 of those letters, removing two due to their focus on new animal drugs and medical devices, respectively.1
The FY24 warning letter total is higher than the 94 letters issued in FY23 and the 74 letters issued in FY22. While the FDA’s top observations remained generally consistent, the FY24 breakdown for both country of origin and facility type varies greatly from previous years.

Non Inspection-Based Warning Letters
Four firms received warning letters for failure to register and list.2 Two of those letters also include inspection-based observations.3
CDER continues to utilize its authority under section 704(a)(4) of the Federal Food, Drug, and Cosmetic Act (FD&C Act) to request documents for remote assessments of drug facilities. In FY24, CDER sent 19 warning letters following 704(a)(4) requests, all of which were issued to international facilities, including facilities in Jordan (1), Malaysia (1), Singapore (1), India (1), Canada (2), Turkey (3), South Korea (3), and China (7). All were directed to facilities producing OTC drug products, including hand sanitizers, toothpastes, and topical analgesics.
The FDA issued 51 non inspection-based warning letters based on website review, one based on product testing, and one based on product label review.4 There were several interesting product categories trending in non-inspection-based warning letters this year that stemmed from website review. The FDA cited manufacturers of unapproved topical lidocaine and other analgesic products used to numb skin for cosmetic procedures such as tattooing, piercing, and laser/dermabrasion (six letters).5 The FDA issued six warning letters to manufacturers of unapproved Semaglutide products6 and six to manufacturers of unapproved topical chemical peel products used for skincare.7 Finally, ammonia smelling salt manufacturers caught the agency’s eye with unapproved drug claims for their products online, which resulted in six letters issued based on website review.8
Key Statistics for Inspection-Based Warning Letters
Two letters included refusal or unreasonable delay in production of documents during an inspection, rendering product manufactured at the facilities adulterated under 21 U.S.C. § 351(j),9 and one letter followed a refusal to allow an FDA inspection.10
Location
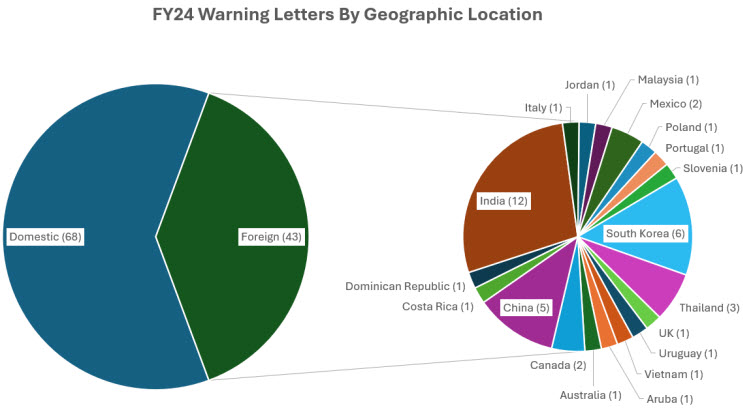
As anticipated, the majority of inspection-based warning letters were issued to domestic firms in FY24. However, we saw a stark contrast in the difference between domestic and international inspections in FY24 and a rise in not only the number but also the diversity in the origin of the letters. In FY23, roughly 83% of inspection-based warning letters were issued to domestic firms (78 of 94). In FY24, roughly 61% of inspection-based warning letters were issued to domestic firms. Specifically, 68 of 111 letters went to domestic firms whereas 43 went to international firms or facilities located outside the U.S.: Aruba (1), Australia (1), Canada (2), China (5), Costa Rica (1), Dominican Republic (1), India (12), Italy (1), Jordan (1), Malaysia (1), Mexico (2), Poland (1), Portugal (1), Slovenia (1), South Korea (6), Thailand (3), United Kingdom (1), Uruguay (1), and Vietnam (1).11
Facilities and Product Categories12
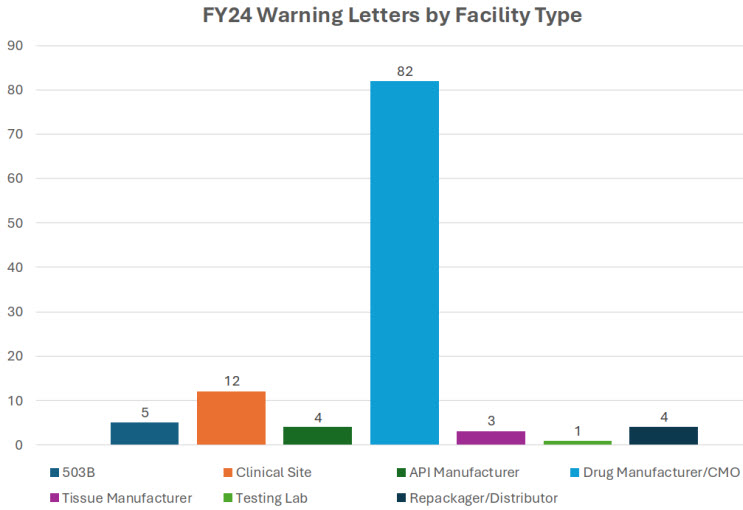
The FDA focused primarily on drug manufacturers in FY24, with fewer 503B warning letters issued than in FY23 and FY22. In FY23, OTC letters made up nearly 35% of all warning letters issued. In FY24, we saw a much higher proportion, with OTC products representing 53% of all inspection-based warning letters. This is certainly a space to watch as the FDA continues to focus its enforcement on these products.

Top Citations
The top five citations look a lot like last year’s, with section 211.100(a) topping the list yet again. The FDA continues to focus on component testing, as various contaminants have permeated the OTC drug space, specifically in hand sanitizers and other topical products.
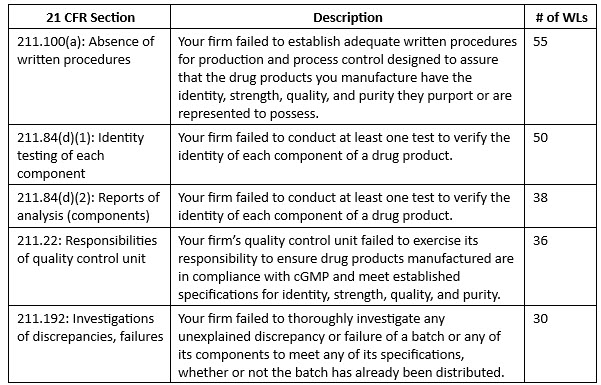
Other notable citations include:
- 21 CFR 211.166(a): Lack of written stability program. The FDA cited 25 firms for failing to ensure the establishment of an adequate ongoing stability program.
- The FDA also cited firms for lack of quality control under section 21 CFR 211.22(a) on 19 separate occasions. This citation, along with 21 CFR 211.22(d) (the failure to establish adequate written procedures applicable to the quality unit and to follow such written procedures, cited in 17 letters), piggyback off of the citations issued under 21 CFR 211.22 in the 36 letters mentioned in the top five. These three citations outline the pervasive trend of inadequate quality control in drug manufacturing.
- The agency also frequently cited firms for lack of release testing (19 letters) under 21 CFR 211.165(a). Here, the firms in question failed to have, for each batch of drug product, appropriate laboratory determination of satisfactory conformance to final specifications for the drug product, including the identity and strength of each active ingredient, prior to release.
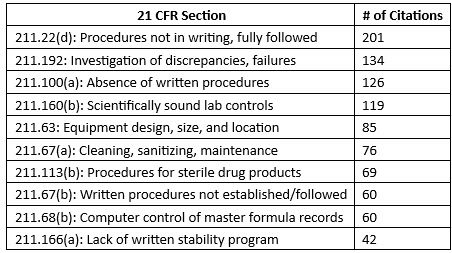
The FDA released data on FDA Form 483 citations issued to each program area in FY24.13 The agency reported 561 483s issued to firms within the drugs program area. While the data is not inclusive of all 483s issued during the fiscal year, the top 10 citations noted on 483s in this space were comparable to those observed in warning letters.
Warning Letter Trends
Component Testing
For the third year in a row, hand sanitizers and other OTC drug products like sunscreen remain a focus for the FDA’s inspection-based warning letters due to their potential to be contaminated with methanol, diethylene glycol (DEG), and/or ethylene glycol (EG). The FDA cites this by stating “your firm failed to test samples of each component for identity and conformity with all appropriate written specifications for purity, strength, and quality (21 CFR 211.84(d)(1)).”
Historically, the FDA has been concerned with the use of ethanol contaminated with methanol to manufacture hand sanitizers as use of such contaminated ingredients has caused lethal poisoning incidents in humans worldwide. This issue peaked after the start of the COVID-19 public health emergency and led to greater FDA focus, resulting in numerous enforcement actions and warning letters.14 This year we saw an uptick in attention on two other contaminants, DEG and EG, which are found in glycerin and propylene glycol used in manufacturing. Like previous findings of methanol contamination, firms cited for this issue typically failed to adequately test each shipment of incoming ingredients/components at risk for contamination. Within the warning letter, the FDA states, “your component identity testing did not include a limit test for diethylene glycol (DEG) and ethylene glycol (EG) on all lots of glycerin before use in the manufacture of your drug products.”15
The FDA includes specific concerns surrounding DEG or EG contamination in 37 of the 111 inspection-based warning letters issued in FY24 and it recommends review of the guidance document, Testing of Glycerin, Propylene Glycol, Maltitol Solution, Hydrogenated Starch Hydrolysate, Sorbitol Solution, and Other High-Risk Drug Components for Diethylene Glycol and Ethylene Glycol, to help meet cGMP requirements when manufacturing drugs containing ingredients at high-risk for DEG or EG contamination.16
Recalls and Import Alerts
In FY24, the FDA mentions recalls in 23 letters. The FDA does not have mandatory recall authority over drug and biologic products. Rather, it relies on regulated industry to initiate voluntary recall actions to remove product from the market and/or warn patients about potential risks.17 The commentary surrounding recalls in FY24 warning letters is more forthcoming and direct than we have seen in past years, especially where the FDA calls out firms for delay or failure to initiate a recall. This is likely a strategy the FDA is employing to persuade voluntary action in a timely manner.
In five instances, the FDA specifically referred to teleconferences wherein it made a specific recommendation that the firm initiate a voluntary recall.18 Four additional warning letters include commentary on the FDA’s requested recalls and the delay or refusal of the company to comply.19 Specifically, the FDA noted that it “made multiple attempts to follow-up with [Aromas Para El Alma S.A.] and [its] registered U.S. Agent. As of the date of this letter, you have not initiated a voluntary recall, nor contacted the Agency after multiple requests to do so.”20 The FDA used its warning letter to Sun Pharmaceuticals Industries Ltd. to chastise them for waiting “over five months to initiate a recall of the affected batches” but also noted they did eventually recall all batches of the adulterated drug product.21 Another example is the delayed recall in Velocity Pharma LLC’s warning letter, where the FDA noted during a discussion that “neither you nor [Kilitch Healthcare India Limited] made a commitment to take market action” and referenced the need to make its own public announcement following the delay in making a recall decision. A voluntary recall was eventually initiated by Kilitch, the CDMO, four months after receipt of the warning letter. In all other instances, the FDA merely acknowledges a company’s recall or commitment to perform a recall.
While import alerts and warning letters appropriately overlap in the enforcement space, we noticed a staggering number of products placed on import alert in FY24.
Three warning letters to sponsors noted a CMO’s presence on an import alert,22 and 27 additional warning letters cited import alerts. Twenty-five of the import alerts noted in the warning letters were for IA 66-40, Detention Without Physical Examination of Drugs From Firms Which Have Not Met Drug GMPs.23 One was for IA 55-03, Detention without physical examination of different forms of heparin and heparin-related products, and one letter was for IA 55-05, Detention without physical examination of finished dosage form drug products, active pharmaceutical ingredients, and inactive ingredients for potentially hazardous microbiological contamination.24
Specific Requests for Warning Letter Response
Warning letters have become more instructive on a point-to-point basis. In addition to clearly articulating why a 483-response failed to address an observation, the FDA is now offering equally instructive guidance on resolution of the issues. The FDA frequently requests specific commitments for each observation with detailed deliverables and timelines.
For example, in response to data integrity lapses cited in the warning letter to Optikem International, the FDA requests a comprehensive investigation into the extent of the inaccuracies in data records and reporting.25 The FDA goes on to state the investigation should include (among other things) a “comprehensive retrospective evaluation of the nature of the testing and manufacturing data integrity deficiencies,” including “[a] current risk assessment of the potential effects of the observed failures on the quality of your drugs. [The] assessment should include analyses of the risks to patients caused by the release of drugs affected by a lapse of data integrity and analyses of the risks posed by ongoing operations.”26 The letter continues, to request “a commitment to have a qualified consultant conduct extensive annual audits, for at least two years, to assist in evaluating CAPA effectiveness after [the firm has] executed [its] data integrity remediation protocol.”27
Some letters also ask for comprehensive risk assessments on certain areas and include a list of elements to be considered and/or included in the assessment. Within these risk assessments, the FDA is specifically directing companies to perform a comprehensive assessment of operations, procedures, personnel, and/or perform audits of systems to identify additional issues. To ensure understanding, the FDA provides website links to relevant guidance documents. The FDA’s detailed instruction on action items needed for remediation are written as if they could be copied and pasted into a contract with a consulting firm.
Accordingly, the consistent trend of recommending consultants has become more instructive. Rather than recommending engagement of a qualified consultant, the FDA now dictates the specific areas of expertise needed by the consultant. For example, the FDA told Sun Pharma Industries they “strongly recommend that [the] firm engage a consultant qualified as set forth in 21 CFR 211.34 to assist your firm in meeting cGMP requirements.”28 Some letters go on to articulate action items. For example, the warning letter issued to Vintage Chemical stated: “The qualified consultant should also perform a comprehensive six-system audit of your entire operation for cGMP compliance and evaluate the completion and efficacy of your corrective actions and preventive actions before you pursue resolution of your firm’s compliance status with the FDA.”29 And, as in years past, in some letters, the FDA also reminds the firm that the “use of a consultant does not relieve your firm’s obligation to comply with cGMP. Your firm’s executive management remains responsible for resolving all deficiencies and systemic flaws to ensure ongoing cGMP compliance.”30
Water Systems
Twenty-four of the 111 inspection-based warning letters specifically noted insufficient control over water systems and/or inadequate water supply, citing ineffective procedures or testing required to ensure acceptability of a water source for use in manufacturing. All 24 letters containing specific concerns surrounding water systems were issued to OTC product manufacturers.
As an example, in its letter to FirstCham Co., Ltd., the FDA stated the firm “failed to adequately design [its] water system to ensure it was suitable for producing water used in the formulation of [its] drug products (i.e., multiple dead-legs, non-circulating loop) and to provide documentation of its qualification. [It] also failed to show that [its] water system is monitored adequately to ensure it consistently produces water that meets appropriate microbial limits.”31
Similarly, in its letter to Prime Lab LLC, the FDA stated that the firm failed to implement “adequate design and procedures for production and process controls for drug products and for maintenance and monitoring of [its] water system used to manufacture drug products, including a lack of investigation into out of limit microbiology test results during the installation of [its] water system (21 CFR 211.100(a) and 211.192)).”32
Conclusion
As we noted at the outset, the FY24 warning letter total is higher than in previous years. Moreover, while the FDA’s top observations remained generally consistent, more letters were issued to international facilities than in the past (and to more varied locations), and more letters were issued to OTC manufacturers. FDA also focused on component testing (as a result of methanol and DEG/EG contamination) and water systems for OTC products. The agency is also becoming more specific and instructive in its letters, and has called out recalls and import alerts more frequently.
Importantly, on October 1, 2024, the FDA implemented a long-planned reorganization of the Human Foods Program and the FDA field operations in response to the Reagan-Udall Foundation Evaluation published in 2022. Specifically, as of October 1, 2024, the Office of Regulatory Affairs (ORA) was renamed the Office of Inspections and Investigations (OII) and compliance staff were relocated to relevant centers to better align the compliance process and enhance collaboration between field investigators and experts at the agency. While this reorganization will not impact the personnel conducting inspections and the timeline of those inspections, the reorganization will impact the internal FDA process for issuing warning letters — ideally streamlining the process and making it more collaborative.
Looking forward to FY25, we will monitor the impact of the reorganization, along with recent reductions in force at the agency, and other changes made in the new Administration on the number of warning letters.
References/Notes
- FDA’s warning letters to Safari Stem Cell, LLC (regarding a new animal drug) and Barkey GmbH & Co. KG (regarding a medical device) were not included in this article.
- Ehy Holdings, LLC, Dr. Sankunni’s Ayurvedic Research Foundation Private Ltd., Zen Enterprises LLC, Media Networks Sydney Pty Limited.
- Inspection-based warning letters issued to Handcock Cosmetics Co., Ltd. (also cited for failure to list product) and Cosmetic Specialty Labs, Inc. (also cited for failure to register and list).
- LightEyez Limited was based on product sample testing and Premier Manufacturing Products, LLC was based on product label review.
- Sky Bank Media, LLC DBA Painless Tattoo Cream Co., TKTX Company, SeeNext Venture, Ltd. Dermal Source, Inc., Indelicare DBA INKEEZE, and Tattoo Numbing Cream Co.
- US Chem Labs, Synthetix Inc. DBA Helix Chemical Supply, Nomida.biz, www.gorillahealing.com, www.semaspace.com, and Ozempen.com.
- Matte Beauty, Repare Skincare, ISIS.GOLD, Skin Beauty Solutions, and Amazon.com, Inc. (issued two letters).
- Native Salts, Innovative Formulations, Spirochaete Research Labs, Skull Smash, Ammonia Sport, and Ward Smelling Salts.
- Regenerative Processing Plant, LLC and S & J International Enterprises Public Company Limited.
- Mei Lan Thailand Co., Ltd.
- The three letters issued to firms located in Puerto Rico are included in the count for domestic warning letters.
- The FY24 Warning Letters by Facility Type table separates Human Cells, Tissues, and Cellular and Tissue-Based Products (HCTP) from other tissue products because three of the letters identified HCTP products as unapproved drugs, where the remaining warning letters issued to tissue product manufacturers were cited under 21 CFR 1271 for poor tissue practices, but are not categorized as unapproved drugs. Thus, the HCTP letters are included in the “drug Manufacturer section of the “Facility Type” table and on their own in the “Product Category“ table. Additionally, in the “Product Category” table, the 21 warning letters in the drug product category refer to prescription drug products as opposed to OTC drug products.
- Inspection Observations, FY 2024 Inspectional Observation Data sets from FDA Form 483, available at https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspection-observations.
- In FY23 and FY22 16 warning letters were sent to firms producing hand sanitizer products.
- See Bell International Laboratories, Inc. letter’s citation of 211.84(d)(1).
- FDA Guidance for Industry, Testing of Glycerin, Propylene Glycol, Maltitol Solution, Hydrogenated Starch Hydrolysate, Sorbitol Solution, and Other High-Risk Drug Components for Diethylene Glycol and Ethylene Glycol, available at https://www.fda.gov/media/167974/download.
- See FDA’s Drug Recalls webpage, https://www.fda.gov/drugs/drug-safety-and-availability/drug-recalls.
- GFA Production Xiamen Co., Ltd, Delsam Pharma, LLC, Seatex, LLC, Global Pharma Healthcare Private Limited, and Laboratorio Magnachem International.
- Aromas Para El Alma S.A., Sun Pharmaceutical Industries Ltd., Velocity Pharma LLC, and Kilitch Healthcare India Limited.
- Aromas Para El Alma, S.A.
- Sun Pharmaceutical Industries Limited.
- Velocity Pharma LLC, EzriCareLLC, and Delsam Pharma LLC.
- IA 66-40, https://www.accessdata.fda.gov/cms_ia/importalert_189.html.
- Mei Lan Thailand Co., Ltd was placed on IA 55-03, https://www.accessdata.fda.gov/cms_ia/importalert_821.html and GFA Production Xiamen Co., Ltd. was placed on IA 55-05, https://www.accessdata.fda.gov/cms_ia/importalert_1126.html.
- Optikem International FDA Warning Letter, available at https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/optikem-international-inc-680264-06202024.
- Id.
- Id.
- Sun Pharmaceutical Industries Limited, FDA Warning Letter, available at https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/sun-pharmaceutical-industries-limited-677337-06182024. Similar language was also included in warning letters to Zydus Lifesciences Limited and Orean Personal Care Ltd., among others.
- Vintage Chemical Inc., FDA Warning Letter, available at https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/vintage-chemical-inc-673930-03272024.
- Warning letters issued to Cosmetics Specialty Labs and Master Paints & Chemicals Corp., among others, include this language.
- FirstCham Co., FDA Warning Letter, available at https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/firstcham-co-ltd-672903-04032024.
- Prime Lab LLC, FDA Warning Letter, available at https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/prime-lab-llc-667057-01022024.
About The Authors:
 Liz Oestreich, J.D., is senior vice president of regulatory compliance at Eliquent Life Sciences (formerly Greenleaf Health). She brings more than nine years of regulatory experience and a diverse background of legal, public policy, and non-profit sector knowledge to her position. As a consultant, Oestreich provides strategic guidance on premarket and postmarket issues to drug, medical device, tobacco, and cannabis companies. She earned her J.D. from the University of the District of Columbia, David A. Clarke School of Law, and her B.A. from the University of Arizona.
Liz Oestreich, J.D., is senior vice president of regulatory compliance at Eliquent Life Sciences (formerly Greenleaf Health). She brings more than nine years of regulatory experience and a diverse background of legal, public policy, and non-profit sector knowledge to her position. As a consultant, Oestreich provides strategic guidance on premarket and postmarket issues to drug, medical device, tobacco, and cannabis companies. She earned her J.D. from the University of the District of Columbia, David A. Clarke School of Law, and her B.A. from the University of Arizona.
 Kalah Auchincloss, J.D., M.P.H., is executive vice president of regulatory compliance and deputy general counsel at Eliquent Life Sciences (formerly Greenleaf Health). She has more than 15 years of food and drug legal, policy, and regulatory experience at the FDA, on Capitol Hill, and in the private sector. At Greenleaf, Auchincloss advises pharmaceutical and medical device companies on compliance, policy, and other regulatory issues. Before moving to Greenleaf, Auchincloss spent six years at the FDA in the Commissioner’s Office and in CDER’s Office of Compliance and Office of Regulatory Policy. She earned her J.D. from Georgetown University, her M.P.H. from Harvard University, and her B.A. from Williams College.
Kalah Auchincloss, J.D., M.P.H., is executive vice president of regulatory compliance and deputy general counsel at Eliquent Life Sciences (formerly Greenleaf Health). She has more than 15 years of food and drug legal, policy, and regulatory experience at the FDA, on Capitol Hill, and in the private sector. At Greenleaf, Auchincloss advises pharmaceutical and medical device companies on compliance, policy, and other regulatory issues. Before moving to Greenleaf, Auchincloss spent six years at the FDA in the Commissioner’s Office and in CDER’s Office of Compliance and Office of Regulatory Policy. She earned her J.D. from Georgetown University, her M.P.H. from Harvard University, and her B.A. from Williams College.
 Erin Hartmann, J.D., is manager of regulatory affairs in Eliquent Life Sciences’ Quality and Compliance Practice. She analyzes FDA regulatory and compliance trends and provides strategic insights to clients. She is a graduate of the University of Texas School of Law and has a B.S. in biology from Case Western Reserve University.
Erin Hartmann, J.D., is manager of regulatory affairs in Eliquent Life Sciences’ Quality and Compliance Practice. She analyzes FDA regulatory and compliance trends and provides strategic insights to clients. She is a graduate of the University of Texas School of Law and has a B.S. in biology from Case Western Reserve University.